
Hola somos Andrea, Miguel y Carlos
Bienvenido. ¿Listo para aprender?
Estamos felices de enseñarte lo que aprendimos en los Clubes de Ciencia México 2021 donde fuimos parte del club 11 "Harness the power of data: Medicine designed by computers".
Empecemos con lo básico, ¿Qué es el docking?
El acoplamiento molecular, también conocido como docking, es un método bioinformático, el cual nos permite predecir y calcular computacionalmente la posición más favorable de interacción entre una proteína y su ligando, en base a sus representaciones tridimensionales (Ballón et al., 2019).
Wow wow, ¡tranquilo cerebrito! ¿Eso qué significa? En pocas palabras, el docking es observar en una computadora cómo se une una molécula a una proteína lo más facil posible.
Esto lo puedes entender con el siguiente ejemplo...
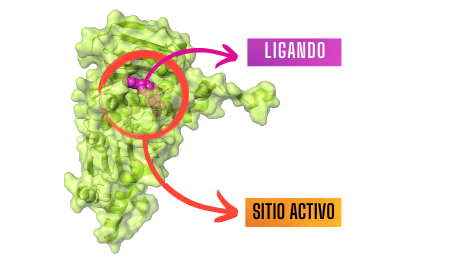
Proteína-Ligando/Automóvil-Conductor
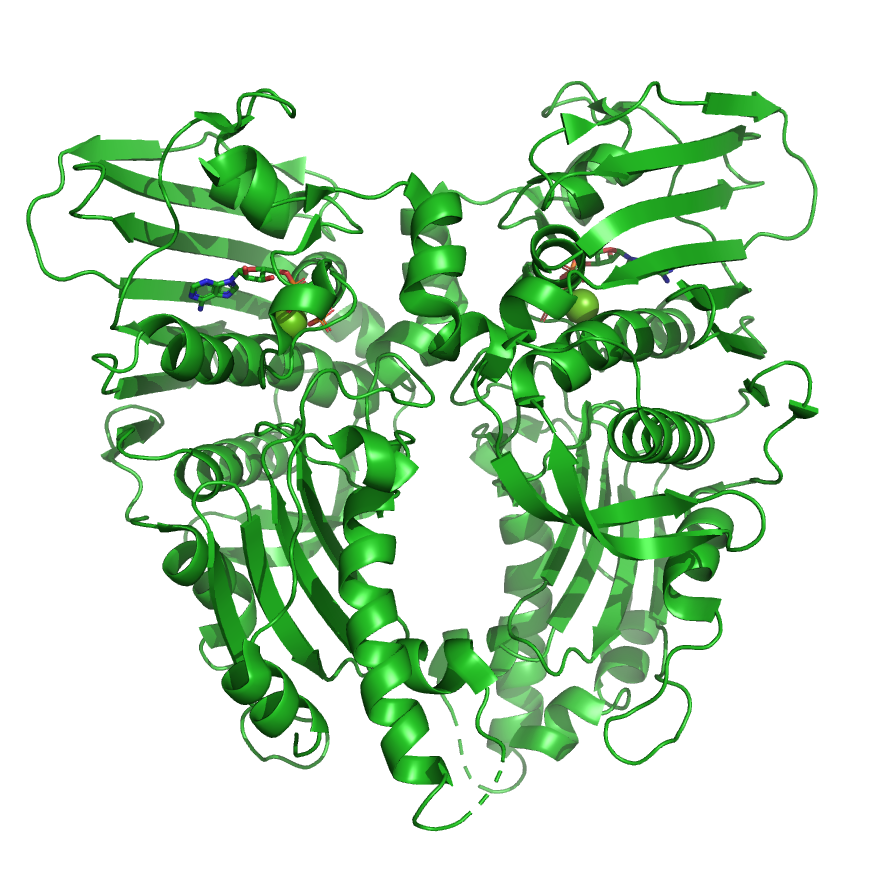
Una proteína es una secuencia de aminoácidos que se unen y se doblan entre sí para formar una estructura 3D. En ellas existe algo llamado sitio activo en donde se llevan a cabo la mayor parte de las interacciones, como la unión con el ligando (una molécula, fármaco, proteína, etc.). El ligando determinará si:
- La proteina funciona normal.
- La proteina funciona mejor.
- La proteina deja de funcionar.
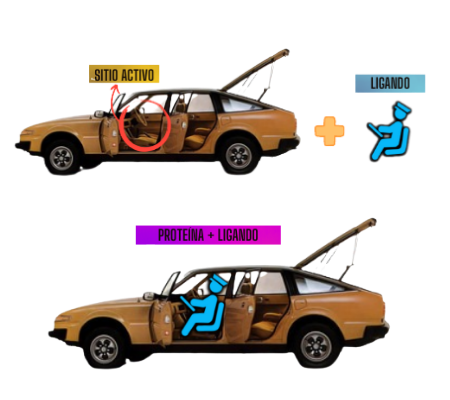
De manera similar, en un automóvil te puedes acomodar de distintas formas en el asiento del conductor, por ejemplo: no podríamos manejar dándole la espalda al volante. ¡Aunque sería muy divertido, la verdad! Bueno, esto tambien pasa con los ligandos y su sitio activo, ya que el ligando se puede acomodar de distintos modos. Sin embargo, el ligando se tendrá que posicionar de una determinada manera para poderse unir con su proteína.
Ahora bien, ¿Cómo el docking se parece a un automóvil y su conductor?
¿Recuerdas el sitio activo de las proteínas? Este es parecido al espacio del conductor de un auto.
Pero, ¿por qué? No te preocupes, a continuación te explicamos...
En este sitio activo se podrá acomodar un ligando, que en este ejemplo será un conductor. El conductor podrá entrar de manera adecuada en el asiento. De igual forma, del conductor dependerá sí el manejo del auto es excelente, bueno o nulo. Esto mismo pasa con las proteínas y su ligando, ya que dependiendo de la unión de estos dos elementos sabremos si la proteína es capaz o no, de funcionar.

Obviamente no todos los conductores manejaran igual de bien un auto ¿verdad? No es lo mismo que maneje Checo Pérez, a que maneje una persona muy distraida. Algo así pasa con los ligandos y las proteínas. Habrán muchos ligandos, por lo que cada uno funcionará de manera distinta en la proteína. En el docking, siempre buscaremos el ligando que funcione de mejor manera con nuestra proteína de interés.
¿Cáncer?
Ahora bien ¿Qué es el cáncer?

El cáncer, es el nombre que se da a un conjunto de enfermedades relacionadas. Esta enfermedad genera que algunas de las células malignas del cuerpo empiecen a dividirse sin detenerse y esparcirse a diferentes tejidos (metástasis). La vida de una célula humana va algo así: crecer y dividirse para formar nuevas células a medida que el cuerpo las necesita, una vez que estas células envejecen o se dañan, mueren y las células nuevas las reemplazan.
Pero ¿qué pasa en las células cancerígenas?
Se podría decir que este tipo de células son muy rebeldes, ya que son la excepeción a este proceso. A medida que las células se hacen más y más anormales, las células viejas o dañadas sobreviven cuando deberían morir, y células nuevas (pero malhechas) se forman cuando no son necesarias. Estas células adicionales pueden dividirse sin interrupción y pueden formar masas que se llaman tumores (Instituto Nacional del Cáncer, 2012).
¿Los tumores son malos?
¡Tristemente, la mayoría de ellos sí! Los tumores cancerosos son malignos y tienen la capacidad de extenderse e invadir a tejidos cercanos. Cuando estos tumores crecen demasiado, desprenden algunas células cancerígenas moviéndose a distintos lugares del cuerpo, formando así, nuevos tumores en lugares distintos al original.
Doxorrubicina.
Una vez que ya hablamos un poco sobre el cáncer, hablemos sobre los medicamentos que se utilizan para combatirlo.
La doxorrubicina (adriamicina) es un antibiótico con actividad antitumoral producido por un pequeño organismo llamado Streptococcus peucetius var. caesius. Debido a su capacidad de roturas sobre cadenas simples y dobles tiene propiedades mutagénicas y carcinogénicas. De manera simple, este antibiótico actúa retardando o deteniendo el crecimiento de las células cancerosas en el cuerpo (American Society of Health, 2012). Este fármaco se utiliza en la actualidad en tratamientos contra el cáncer debido a su alta efectividad (Carranza, 2013). Pero claro, no todo puede ser bonito ¿Verdad? La realidad es que este antibiótico suele ser muy tóxico, pudiendo generar mucho daño en los ovarios y las células de los pacientes (Salih, et. al. 2015).
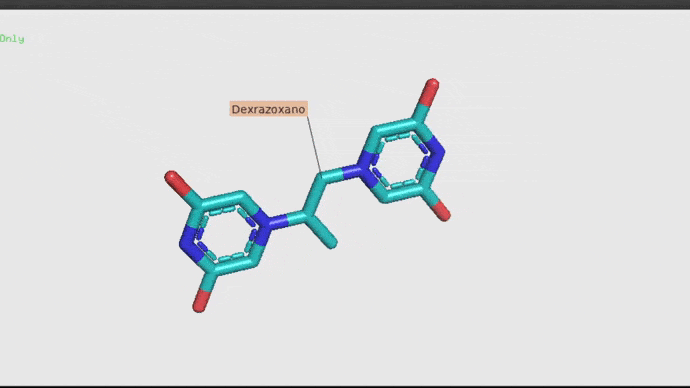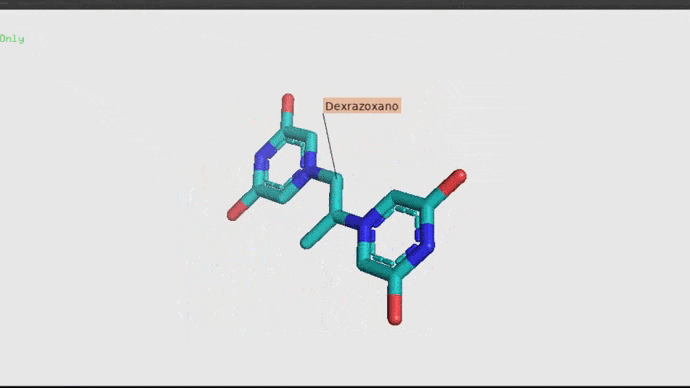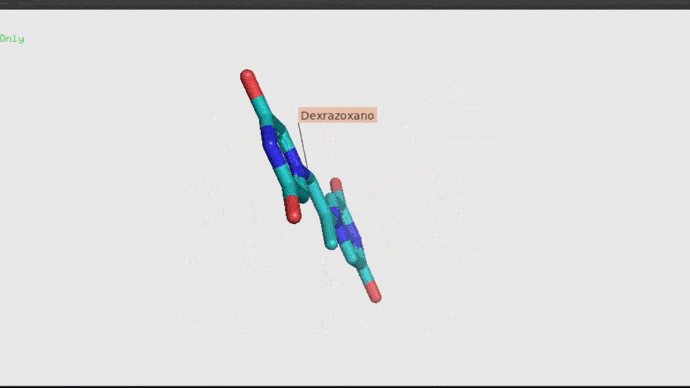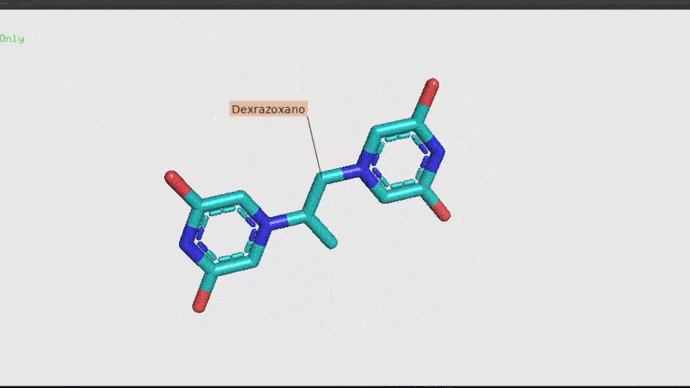
Dexrazoxano.
Tal vez y ahora estés pensando ¿No existe algún medicamento que pueda servir contra el cáncer sin generar tanto daño?
Tenemos una excelente noticia para ti... ¡SÍ!
El antibiótico Dexrazoxano es una alternativa innovadora, el cual se ha demostrado que previene la toxicidad ovárica. Funciona como quimioprotector y se utiliza para reducir los efectos indeseados producidos por las quimioterapias (Salih, et. al. 2015). Así también, el dexrazoxano actúa como protector del corazón a los efectos nocivos de la quimioterapia. El dexrazoxano es un potente 'agente quelante intracelular', en palabras simples, esto significa que “secuestra” iones metálicos del cuerpo, como los radicales libres nocivos, cuidando así al corazón de posibles daños (OncoLink Team, 2019).
Topoisomerasa II
La pregunta del millón ¿Por qué la Topoisomerasa II? ¿Acaso es muy especial?
Como la topoisomerasa II participa en la multiplicación y el crecimiento de las células (se encarga de cortar las dobles hélices y dar giros a las cadenas de ADN), el bloqueo de la actividad de esta enzima puede retardar o detener el crecimiento de células cancerosas. Es aquí donde entran tanto la doxorrubicina como el dexrazoxano, ya que estos dos ligantes son inhibidores, esto significa que impiden la actividad de la enzima topoisomerasa II (Instituto Nacional del Cáncer, 2012).
Por eso, nos dimos a la tarea de comparar y analizar dichos inhibidores para saber cuál podría funcionar mejor a la hora de aplicarlo, llevando a cabo un docking molecular.
¿Quieres hacer docking con nosotros?
A continuación te invitamos a que realices un docking con nosotros. Tranquilo, no te preocupes, nosotros te guiaremos por este asombroso proceso.
Estas son algunas cosas que necesitarás para poder realizar este docking.
Pymol
Este es un visualizador molecular el cual te permitirá conocer más sobre la estructura de tus moléculas.
Click aqui para descargar
Tutorial de descarga
Autodock Tools
AutoDock Tools es un conjunto de herramientas de acoplamiento automatizadas.
Click aqui para descargar
Tutorial de descarga
LigPlot
Generador de representaciones esquemáticas en 2D para complejos proteína-ligando.
Click aqui para descargar
Tutorial de descarga
¿Todo listo? Empecemos
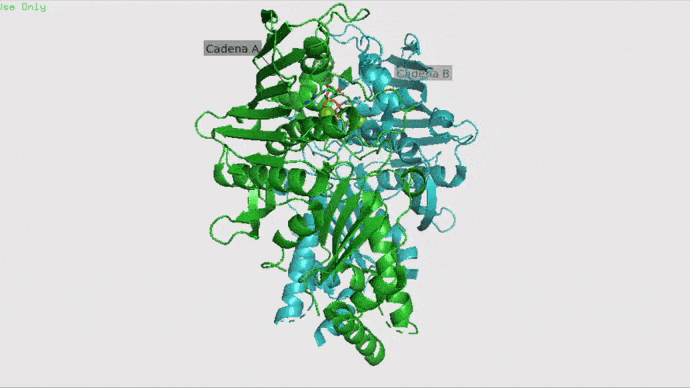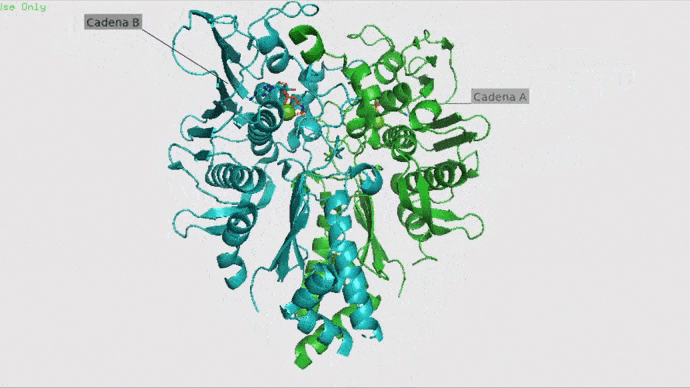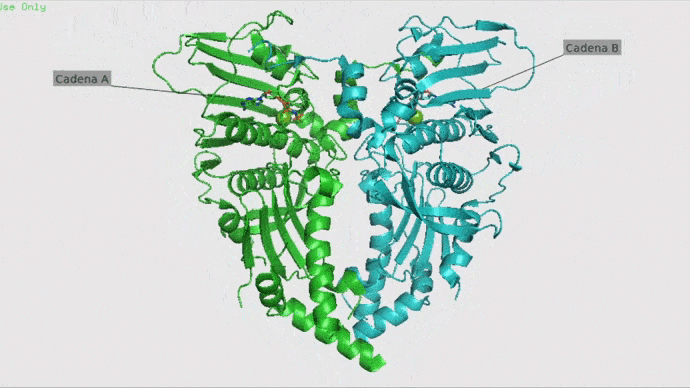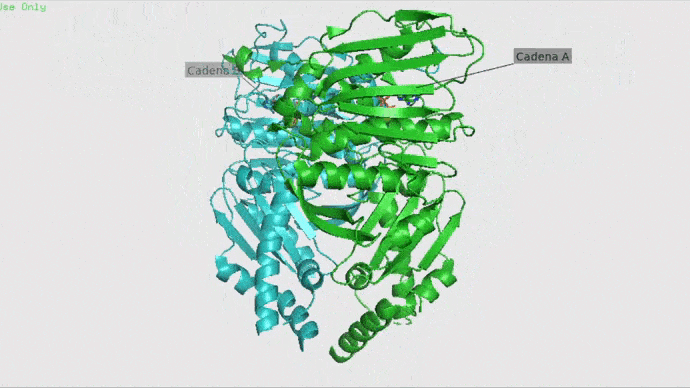
Paso 1. Pymol
Primero abriremos Pymol, este programa nos permitirá visualizar y preparar las moléculas con las que vamos a trabajar, es decir, preparar nuestra proteína y el ligando por separado. En nuestro caso, usaremos la “cadena A” de la Topoisomerasa II como proteína y como ligandos utilizaremos ANP (ligante natural), DM2 (Doxorrubicina) y CDX (Dexrazoxano). Si quieres puedes usar nuestros archivos:
Carpeta con archivos descargables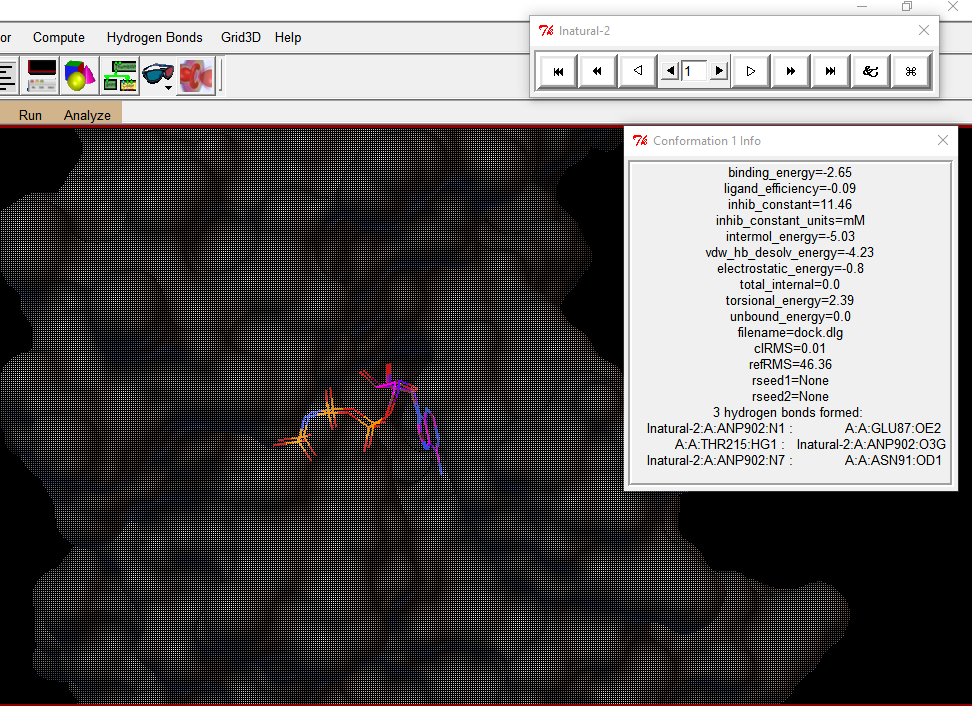
Paso 2. AutodockTools
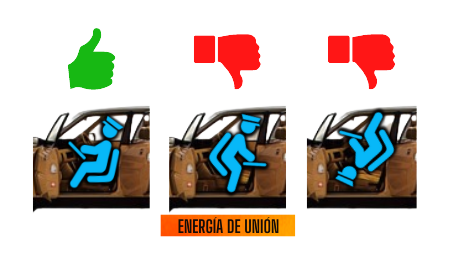
Cuando tenemos nuestras moléculas listas, es tiempo de UNIRLAS! Para ello, abriremos Autodock Tools y haremos una secuencia de pasos que nos permitirán realizar el docking molecular. Tranquilo, esto puede tardar unos minutos, mientras puedes tomar un café o tu bebida favorita! Una vez que se haya realizado el docking Autodock te dará 10 acoplamientos, tendrás que seleccionar al mejor (físicamente hablando); dicha decisión podrás hacerla observando las energías de unión en cada posibilidad, aquel con la MENOR energía, será el mejor docking.
¿Quieres ver todos los pasos a detalle?
Pasos para hacer un docking
Después de haber instalado correctamente AutodockTools (ADT) haremos lo siguiente:
1- Crear un "workfolder" o "carpeta" para archivos del trabajo y ponerle el nombre que tú desees y ubicarlo de preferencia en el Disco (C:) (Ejemplo: docking_CdeCMx)
2- Abrir el programa ADT
Una vez abierto, deberás seleccionar tu directorio, o sea, elegir la carpeta que el programa leerá de ahora en adelante:
a) Abre ADT> File> Preferences>set> Startup Directory (aquí eliges tu carpeta del directorio colocando su ruta). Por ejemplo: “C:\docking_CdeCMx”
3- Prepara tu ligando y proteína en formato ".pdb". Si lo hiciste anteriormente en PyMol, ¡perfecto!
Ahora puedes cambiar el nombre para diferenciarlos mejor, como “proteina.pdb” y “ligando.pdb”
Ahora sí, puedes empezar a hacer el docking como la Ciencia manda!
1- Abre ADT:
Selecciona la pestaña File > Read Molecule> Select Protein File (Aquí eliges el archivo de “proteina.pdb”)
Después:
Edit > Hydrogens >Add>>>>>> Polar Only >OK
Edit > Hydrogens> Merge Non Polar > Continue (si aparece una ventana emergente, solo haz click en "continue")
Luego:
Edit > Charges > Add Kolman Charges> Ok
File > Save > Write PDB> >Sort Nodes (Check) > OK > (Overwrite) YES
Bueno! En el siguiente paso debes intentar preparar el ligando:
Ligand > Input > Open> Select Ligand File (“.pdb” file) > OK
Ahora vuelve a repetir los pasos: (en esta sección deberás agregarle carga a tu ligando).
Para hacer esto tendrás que dar Click en el menú de Editar, después:
Edite> Charges> compute Gasteiger
Ligand > Output > Save as PDBQT
******************************
Ahora deberás ejecutar AutoGrid4:
Para correr el AutoGrid sigue los siguientes pasos:
1-Grid > Macromole > Choose> Click (Protein) > Select Molecule > OK > save
2-Grid> Set Map types >Choose Ligand> Click (Ligand) > Select Ligand
3-Grid > GridBox> Set the BOX> File > Close Saving Current
4-Grid > Output > Save GPF> grid.gpf> save
5-Run > Run AutoGrid:
a) Selecciona la ruta del programa: ‘autogrid4.exe’ (i.e. C:\Program Files (x86) \The Scripps Research Institute\Autodock\4.2.6)
b) Selecciona el archivo con los parámetros: ‘grid.gpf’ (Deberás seleccionarlo dependiendo de en dónde está tu carpeta de trabajo)
c)¡Arrancalo!
Nota: en este paso deberás esperar a que el programa ADT responda. ¡Por favor no deshagas ningún cambio!
***************************************************************
Una vez que hayas realizado estos pasos tendrás que:
Run AutoDock Tools:
1)Docking > Macromolecule > Set Rigid filename> Select ‘Protein.pdbqt’ > Open
2-Docking > Ligand > Choose> Click ‘Ligand’ > Select Ligand> Accept
3-Docking > Search Parameters > Genetic Algorithm> Accept
4-Docking > Output > Lamarckian GA> Save file as ‘dock.dpf’
5-Run > Run AutoDock
a) Selecciona la ruta del programa: ‘autodock4.exe’. Esta dependerá de la ubicación donde hayas descargado ADT. Por ejemplo: (C:\Program Files (x86)\The Scripps Research Institute\Autodock\4.2.6\autodock4.exe)
b) Selecciona el archivo : ‘dock.dpf’ (Basado en la ubicación de tu carpeta de trabajo)
c) Launch!
Espera a que acabe, no cierres el programa.
***********************************************************
Docking result analysis:
Análisis de los resultados del docking.
1-Analyze > Docking > Open> Select ‘dock.dlg’ > Open >Assign Ligand New Name > OK
2-Analyze > Macromolecule > Choose> Click ‘Protein’ > Select Macromolecule
3- Analyze > Conformations> Play, Ranked By Energy > Click on the ‘&’ Button
4-Set Play Options >Check ‘Build H-Bonds’> View the hydrogen bonds formed > Check ‘Show Info’>
View the Interaction Energy > Build Current Write Complex> Save as ‘Result.pdb’ ” Save....
¡Ahora podrás observar tus resultados!
Paso 3. LigPlot
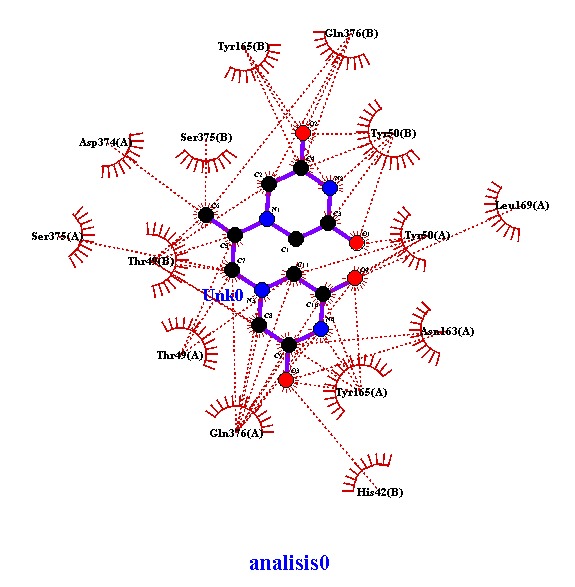
Debido a que el análisis solo se puede realizar con un acoplamiento será importante seleccionar uno. Un tip que te podemos dar es: siempre el archivo 0 será el que tenga una menor energía de unión, por lo cual, será el mejor docking. El análisis del docking es sumamente importante ya que este nos ayudará a saber cómo se comportan nuestros ligandos y cómo interactúa con la proteína de interés.
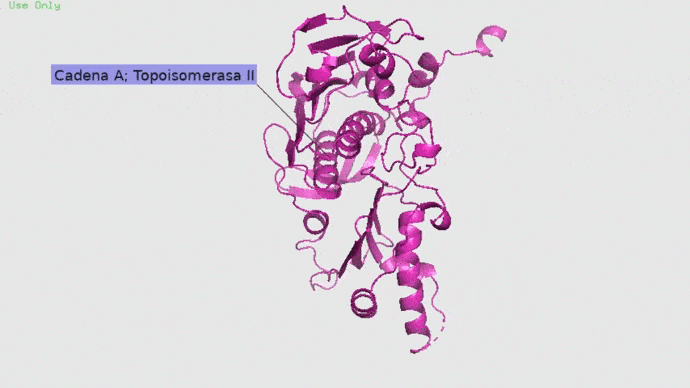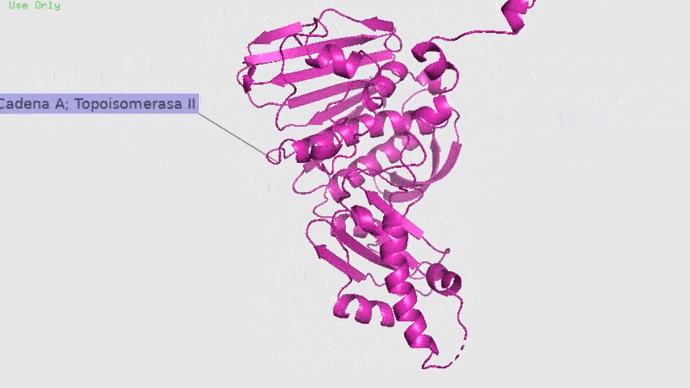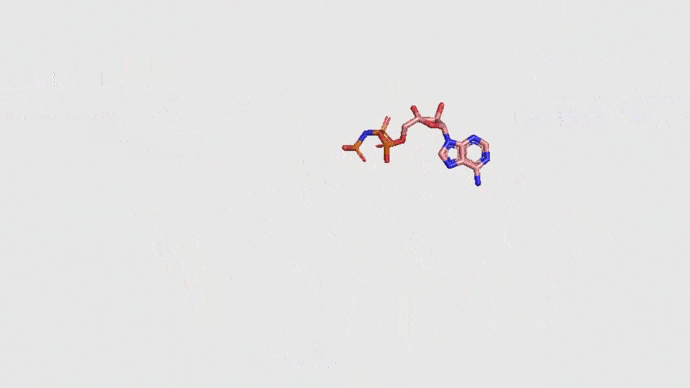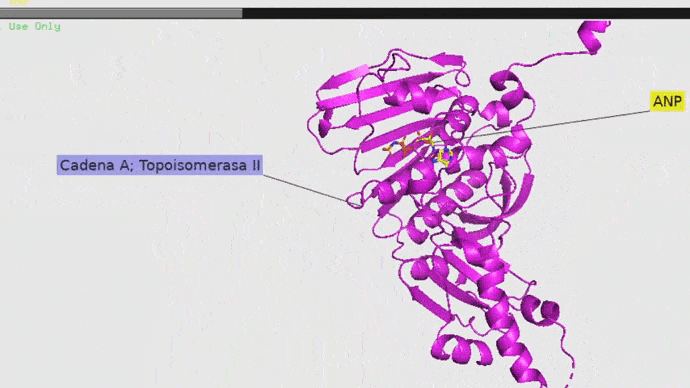
Resultados del docking con Ligante Natural (ANP)
AutodockTools: Para empezar, en el primer docking (ligante natural), la energía de unión que resultó para el mejor de los acoplamientos fue -2.65 kcal/mol , lo que quiere decir que la unión entre la Topoisomerasa II tiene algo de afinidad para permitir ese acople según los resultados de Autodock Tools.
LigPlus: Al visualizar los datos en LigPlus, se resalta la molécula del ligando ANP (magenta) y todos los puentes de H (rojo punteado) que tiene con cada uno de los aminoácidos que la rodean. Además, se establecieron interacciones muy cercanas y consistentes, suficientes para pensar que gracias a ellas la unión proteína-ligando es estable y espontánea, éstas surgen a partir de los aminoácidos marcados en color verde, específicos para cada análisis (Acoplamiento 0, Acoplamiento 5 y Acoplamiento 9), para así recabar los siguientes datos:
Tabla 1.Resultados LigPlus; Cadena A-ANP
| # Acoplamiento | Aminoácido | Distancia |
|---|---|---|
| Acoplamiento 0 | Glu87 Asp94 Ser149 Lys168 Thr215 |
2.73 å 3.12 å 2.92 å 3.13 å 3.68 å |
| Acoplamiento 5 | Glu87 Asp94 Ser149 Lys168 Thr215 |
2.31 å 3.03 å 3.33 å 3.26 å 2.52 å |
| Acoplamiento 9 | Tyr50 | 2.78å |
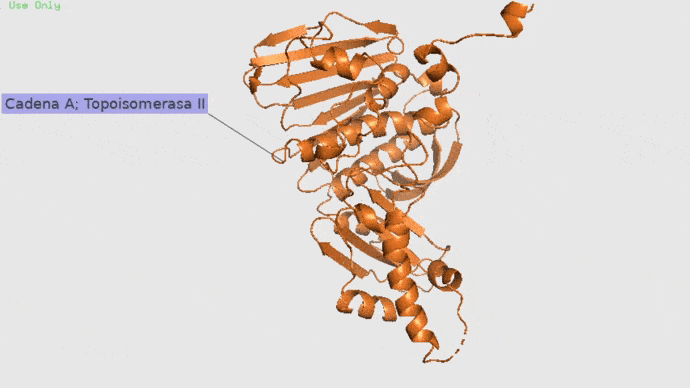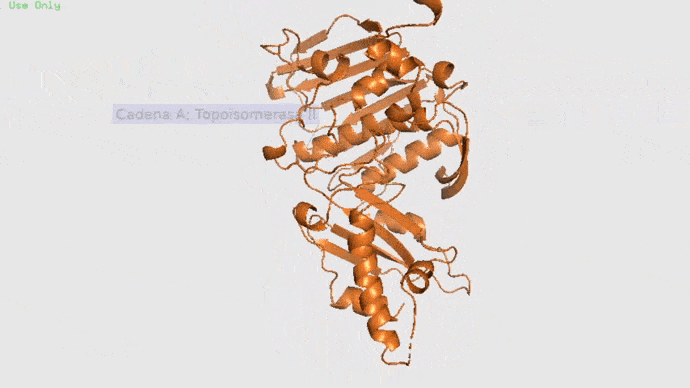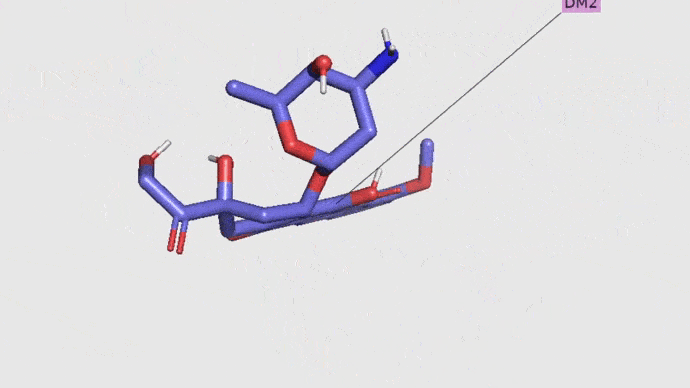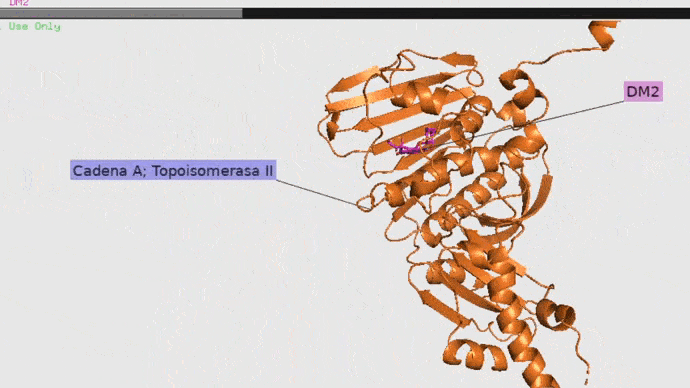
Resultados del docking con Doxorrubicina
AutodockTools: Para empezar, en el primer docking (ligante natural), la energía de unión que resultó para el mejor de los acoplamientos fue -6.76 kcal/mol, lo que quiere decir que la unión entre la Topoisomerasa II tiene algo de afinidad para permitir ese acople según los resultados de Autodock Tools.
LigPlus: Al visualizar los datos en LigPlus, se resalta la molécula del ligando DM2 (magenta) y todos los puentes de H (rojo punteado) que tiene con cada uno de los aminoácidos que la rodean. Además, se establecieron interacciones muy cercanas y consistentes, suficientes para pensar que gracias a ellas la unión proteína-ligando es estable y espontánea, éstas surgen a partir de los aminoácidos marcados en color verde, específicos para cada análisis (Acoplamiento 0, Acoplamiento 4 y Acoplamiento 9), para así recabar los siguientes datos:
Tabla 2.Resultados LigPlus; Cadena A-DM2
| # Acoplamiento | Aminoácido | Distancia |
|---|---|---|
| Acoplamiento 0 | Asn91 Asn95 Asn120 Ile141 Thr147 Lys168 Thr215 |
2.47 å 3.13 å 3.24 å 2.72 å 2.40 å 2.69 å 3.03 å |
| Acoplamiento 4 | Asn120 Lys123 Ile125 Ser149 Thr215 |
2.46 å 3.00 å 2.73 å 3.11 å 2.69 å |
| Acoplamiento 9 | Arg98 Thr215 |
2.53 å 2.73 å |
Resultados del docking con Dexrazoxano
AutodockTools: Para empezar, en el primer docking (ligante natural), la energía de unión que resultó para el mejor de los acoplamientos fue -5.00 kcal/mol, lo que quiere decir que la unión entre la Topoisomerasa II tiene algo de afinidad para permitir ese acople según los resultados de Autodock Tools
LigPlus: Al visualizar los datos en LigPlus, se resalta la molécula del ligando CDX (magenta) que tiene con cada uno de los aminoácidos que la rodean. Además, se establecieron interacciones muy cercanas y consistentes, suficientes para pensar que gracias a ellas la unión proteína-ligando es estable y espontánea, éstas surgen a partir de los aminoácidos marcados en color verde, específicos para cada análisis (Acoplamiento 0, Acoplamiento 5 y Acoplamiento 9), para así recabar los siguientes datos:
Tabla 3.Resultados LigPlus; Cadena A-CDX
| # Acoplamiento | Aminoácido | Distancia |
|---|---|---|
| Acoplamiento 0 | Ninguno relevante | --- å |
| Acoplamiento 4 | Ninguno relevante | --- å |
| Acoplamiento 9 | Tyr50 | 2.78 å |
Conclusiones
Dockings
Al término del proyecto se obtuvo exitosamente el acoplamiento de la Topoisomerasa II humana con tres ligandos: ANP, MD2 y CDX.
Energías de Unión
Cada acoplamiento arrojó energías diferentes, donde la "mejor energía" fue para DM2 (-6.75 kcal/mol), posteriormente CDX (-5.00 kcal/mol) y finalmente ANP (-2.65 kcal/mol).
Tratamiento Cáncer
Tanto DM2, como CDX mostraron potencial de inhibición, sin embargo, CDX sería el mejor candidato a ser usado como tratamiento debido a su bajo riesgo en complicaciones por efectos secundarios.
Galeria de resultados
A continuación se muestran las imagenes obtenidas en el docking.
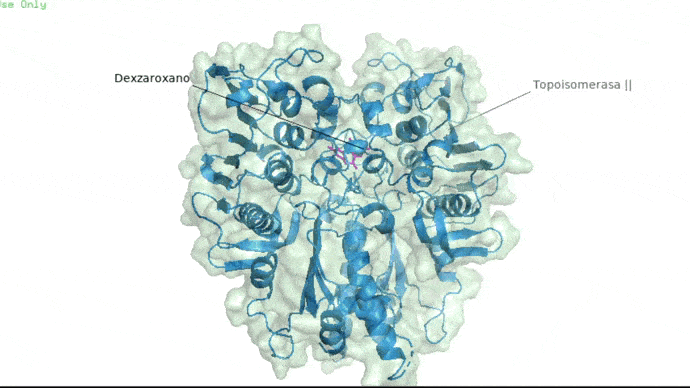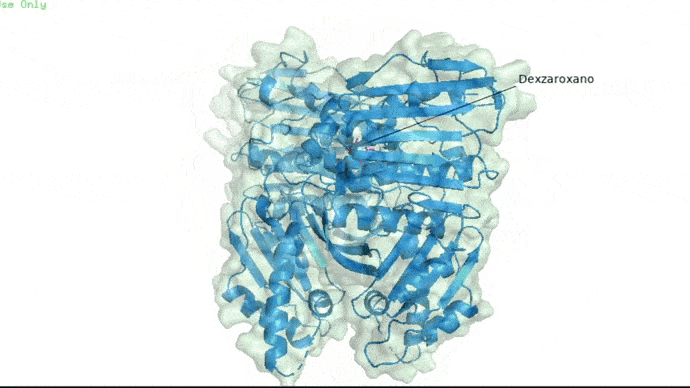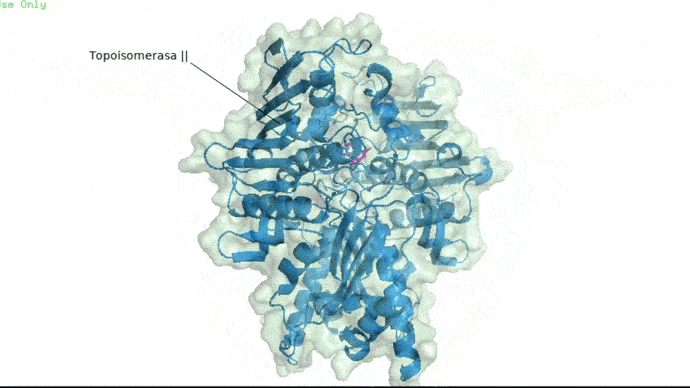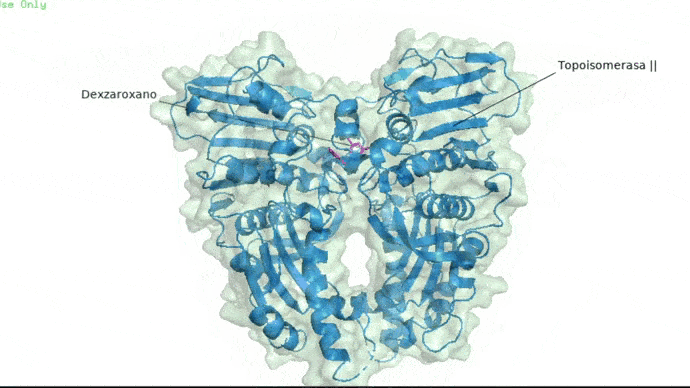
Topoisomerasa
Topoisomerasa II con cadena A y B con su ligando natural. Imagen obtenida en Pymol.
Dexrazoxano
Ligando dexrazoxano, inhibidor de la toposiomerasa II. Imagen obtenida en Pymol.
Doxorrubicina
Ligando doxorrubicina, inhibidor de la toposiomerasa II. Imagen obtenida en Pymol.
ANP en topoisomerasa II
Se muestra al ligando natural (ANP) unido a la topoisomerasa II mediante el docking molecular. Imagen obtenida en Pymol.
Dexrazoxano en topoisomerasa II
Se muestra al ligando Dexrazoxano unido a la topoisomerasa II mediante el docking molecular. Imagen obtenida en Pymol.
Doxorrubicina en topoisomerasa II
Se muestra al ligando Doxorrubicina unido a la topoisomerasa II mediante el docking molecular. Imagen obtenida en Pymol.
Análisis 0 de docking con Dexrazoxano
Se muestra el analisis del acoplamiento 0 de Dexrazoxano con la topoisomerasa II. Se muestran las interacciones hidrofóbicas y no cuenta con puentes de hidrógeno. Imagen obtenida en LigPlot.
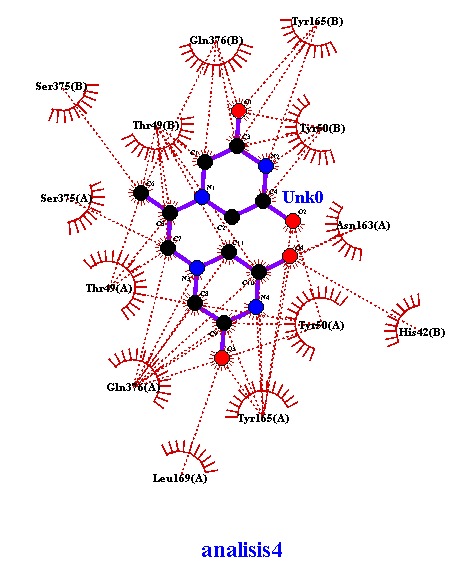
Análisis 4 de docking con Dexrazoxano
Se muestra el analisis del acoplamiento 4 de Dexrazoxano con la topoisomerasa II. Se muestran las interacciones hidrofóbicas y no cuenta con puentes de hidrógeno. Imagen obtenida en LigPlot.
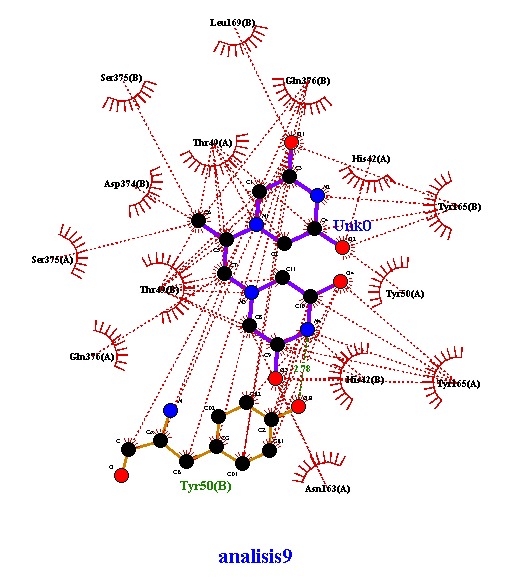
Análisis 9 de docking con Dexrazoxano
Se muestra el analisis del acoplamiento 9 de Dexrazoxano con la topoisomerasa II. Se muestran las interacciones hidrofóbicas, sí cuenta con puentes de hidrógeno. Imagen obtenida en LigPlot.
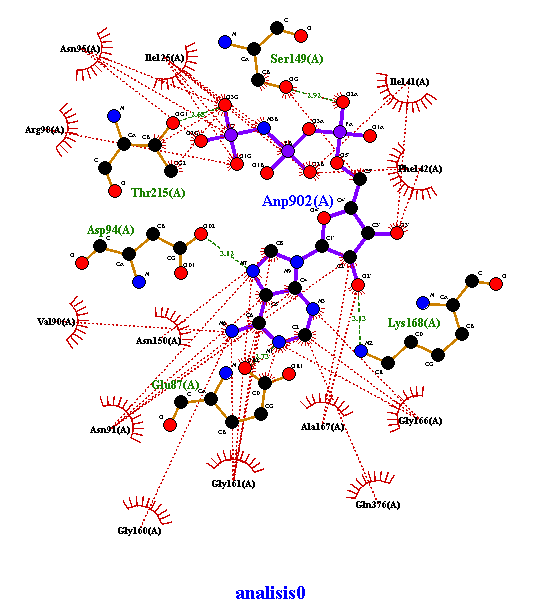
Análisis 0 de docking con ANP
Se muestra el analisis del acoplamiento 0 del ANP con la topoisomerasa II. Se muestran las interacciones hidrofóbica, sí cuenta con puentes de hidrógeno. Imagen obtenida en LigPlot.
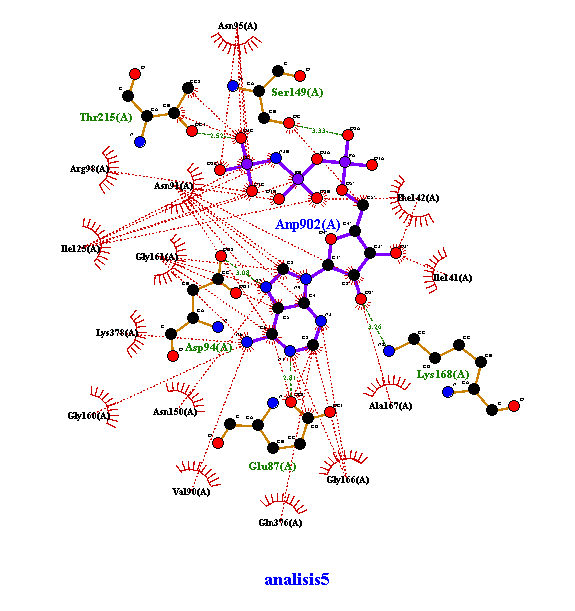
Análisis 5 de docking con ANP
Se muestra el analisis del acoplamiento 5 de ANP con la topoisomerasa II. Se muestran las interacciones hidrofóbicas, sí cuenta con puentes de hidrógeno.. Imagen obtenida en LigPlot.
Análisis 9 de docking con ANP
Se muestra el analisis del acoplamiento 9 de ANP con la topoisomerasa II. Se muestran las interacciones hidrofóbicas, sí cuenta con puentes de hidrógeno. Imagen obtenida en LigPlot.
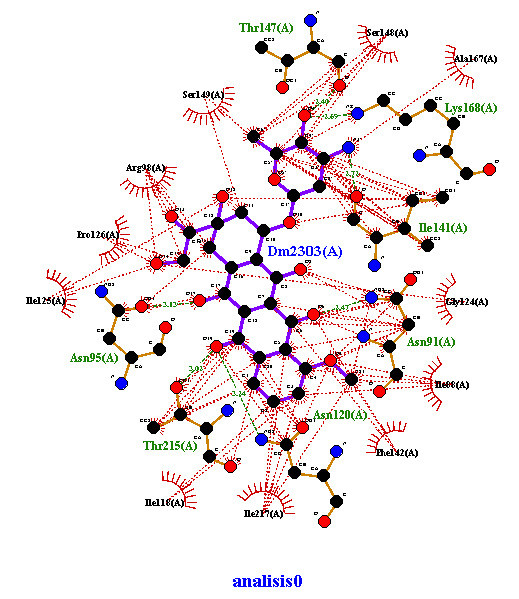
Análisis 0 de docking con Doxorrubicina
Se muestra el analisis del acoplamiento 0 de Doxorrubicina con la topoisomerasa II. Se muestran las interacciones hidrofóbicas, sí cuenta con puentes de hidrógeno. Imagen obtenida en LigPlot.
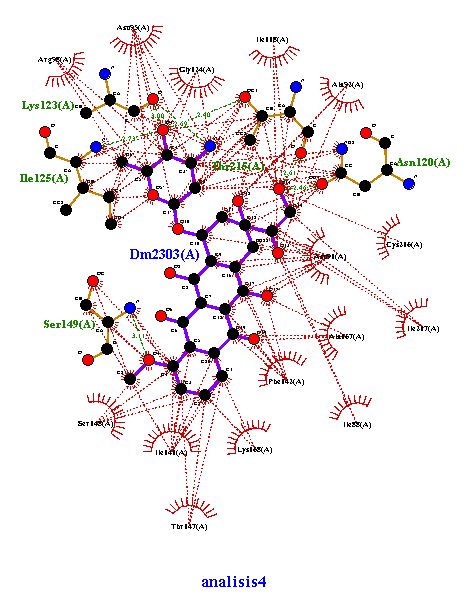
Análisis 4 de docking con Doxorrubicina
Se muestra el analisis del acoplamiento 4 de Doxorrubicina con la topoisomerasa II. Se muestran las interacciones hidrofóbicas, sí cuenta con puentes de hidrógeno. Imagen obtenida en LigPlot.
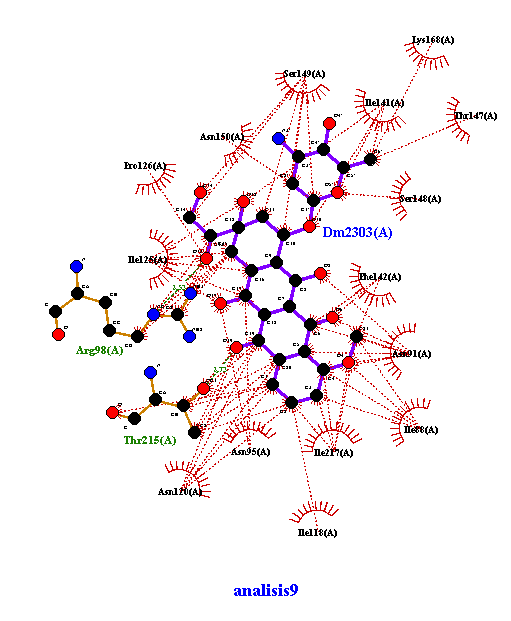
Análisis 9 de docking con Doxorrubicina
Se muestra el analisis del acoplamiento 4 de Doxorrubicina con la topoisomerasa II. Se muestran las interacciones hidrofóbicas, sí cuenta con puentes de hidrógeno. Imagen obtenida en LigPlot.
Testimonios

Andrea Zaragoza Rodríguez
Estudiante de Biotecnología
Me gustó mucho participar en esta edición del CdCMx, agradezco mucho la oportunidad de haber sido parte de esta edición. Considero que el conocimiento de técnicas como el Docking es vital en una disciplina como lo es la biotecnología, ya que nos permite conocer más sobre el modo de acción de distintas proteínas lo cual nos permitirá utilizarlas de una manera más efectiva, generando así, una variedad de productos de interés para la población como pueden ser los fármacos. Sin duda alguna esta información me será muy útil en el futuro. ¡Gracias!

Miguel Olmos Galarza
Estudiante de Ing. en Materiales
Estoy muy feliz de participar en esta edición de CdCMx y mucho más en el club que me tocó, porque abordé temas de la medicina muy interesantes y en los que me quiero especializar. El docking es una estrategia muy útil para tener nuevos avances en los fármacos, Como un ingeniero en materiales, me alegra más comprender este proceso, debido a que estaré también encargado del desarrollo in vivo del tratamiento. Agradezco a mis instructores y compañeros que lograron un club muy increíble.

Carlos Amaro Osorio
Estudiante de Biotecnología
Me voy feliz como una lombriz. Al haber realizado los dockings moleculares se logró no solo obtener información bioinformática de relevancia médica, económica y científica, si no, en lo personal puedo decir que adquirimos los conocimientos básicos y generales sobre la importancia y la aplicación que tiene hoy en día el docking. Sin duda alguna, el desarrollo y trabajo con este tipo de herramientas será una promesa para el futuro en la búsqueda de nuevos posibles tratamientos. ¡Gracias @ClubesdeCienciaMéxico!
Nuestro equipo
Carlos Amaro Osorio
Biotecnología
Miguel Olmos Galarza
Ing. en Materiales
Andrea Zaragoza R.
Biotecnología
Clubes de Ciencia
Edición 2021Video del proyecto
¡En estos pocos minutos, resumimos la experiencia y resultados que nos llevamos de este asombroso club!
¡Dale un vistazo!